Making Canonical Workflow Building Blocks interoperable across workflow languages
Cite as
Stian Soiland-Reyes, Genís Bayarri, Pau Andrio, Robin Long, Douglas Lowe, Ania Niewielska, Adam Hospital, Paul Groth (2022):
Making Canonical Workflow Building Blocks interoperable across workflow languages.
Data Intelligence 4(2)
https://doi.org/10.1162/dint_a_00135
Making Canonical Workflow Building Blocks interoperable across workflow languages
Stian Soiland-Reyes¹², Genís Bayarri³, Pau Andrio⁴, Robin Long⁵⁶, Douglas Lowe⁶, Ania Niewielska⁷, Adam Hospital³, Paul Groth²
¹ Department of Computer Science, The University of Manchester, Manchester, UK
² Informatics Institute, University of Amsterdam, Amsterdam, The Netherlands
³ Institute for Research in Biomedicine (IRB Barcelona), The Barcelona Institute of Science and Technology (BIST), Barcelona, Spain
⁴ The Spanish National Bioinformatics Institute (INB), Barcelona Supercomputing Center (BSC), Barcelona, Spain
⁵ Lancaster University, Lancaster, UK
⁶ Research IT, The University of Manchester, Manchester, UK
⁷ European Bioinformatics Institute (EMBL-EBI), Cambridge, UK
Abstract
We introduce the concept of Canonical Workflow Building Blocks (CWBB), a methodology of describing and wrapping computational tools, in order for them to be utilised in a reproducible manner from multiple workflow languages and execution platforms. The concept is implemented and demonstrated with the BioExcel Building Blocks library (BioBB), a collection of tool wrappers in the field of computational biomolecular simulation. Interoperability across different workflow languages is showcased through a protein Molecular Dynamics setup transversal workflow, built using this library and run with 5 different Workflow Manager Systems (WfMS). We argue such practice is a necessary requirement for FAIR Computational Workflows and an element of Canonical Workflow Frameworks for Research (CWFR) in order to improve widespread adoption and reuse of computational methods across workflow language barriers.
1. Introduction
The need for reproducibility of research software usage is well established [1,2,3], and adaptation of workflow management systems (WfMS) together with software packaging and containers [4] have been proposed as key ingredients for making research software usage FAIR and reproducible [5,6,7]. Recently it is also argued that computational workflows should also be treated as FAIR Digital Objects [8] in their own right, with identifier, metadata [2] and interoperability requirements [9].
BioExcel, a European Centre of Excellence for Computational Biomolecular Research, has a particular focus on the research domains molecular dynamics simulations and bioinformatics with use of High Performance Computing (HPC) to approach Exascale performance, while also improving usability. The BioExcel Building Blocks (BioBB) [10] have been created as portable wrappers of open-source computational tools identified as useful for BioExcel workflows, forming several families of documented and interoperable operations that can be called from multiple workflow systems. This interoperability is shown with the BioBB demonstrator workflows, along with multiple tutorials and notebooks.
We propose that these building blocks and their families can themselves be considered composite Digital Objects: collections of software packages and their source code, guides and tutorials, as well as workflow management system integrations and workflow examples. In addition, the building blocks, as wrappers of upstream open source tools, benefit from and refer to the tools’ existing documentation, support forums, academic publications and wider development context.
Given BioBB as a starting point, we define a generalized methodology of Canonical Workflow Building Blocks (CWBB), through the definition of a set of requirements and recommendations for how to formalize and develop a family of compatible computational tools as Digital Objects. These building blocks let researchers instantiate a Canonical Workflow in multiple workflow management systems, while also benefiting from the FAIR aspects of the CWBB Digital Objects.
2. Methods
The BioExcel Building Blocks library [10], created and implemented within the BioExcel CoE, is a collection of portable wrappers of common biomolecular simulation tools. The BioBB library is designed to i) increase the interoperability between the tools wrapped; ii) ease the implementation of biomolecular simulation workflows; and iii) increase the reusability and reproducibility of the generated workflows. To achieve these main goals, the library was designed following the FAIR principles for research software development best practices [7].
The result is a collection of building block modules, divided in sets of tool wrappers focused on similar functionalities (e.g. Molecular Dynamics, Virtual Screening). Each of the modules is built from a combination of (i) software packaging (Pip, BioConda, BioContainers), (ii) documentation (ReadTheDocs), (iii) interactive tutorials (Jupyter Notebooks, Binder), (iv) registry & findability (bio.tools, BioSchemas, WorkflowHub), (v) WfMS integration stubs (CWL, Galaxy, PyCOMPSs), (vi) source Code (GitHub) and (vii) REST APIs (OpenAPI, Swagger). Notably all building blocks follow the same pattern of installation, configuration and interaction.
Since the publication of the library, several new building block modules (Chemistry, Machine Learning, AMBER MD, Virtual Screening, etc.) have been added, and the set of operations for the existing BioBB families have been expanded. While we previously provided curated adapters (Figure 1) for running BioBB in workflow systems using Common Workflow Language (CWL) and PyCOMPSs, along with Galaxy Toolshed bindings, we have now started auto-generating these bindings, along with command line wrappers and REST web service APIs, using annotations within BioBB’s Python docstrings as source. These annotations include sufficient information for a WfMS to launch a particular building block: input and output parameters (including mandatory/optional flags), compatible formats (including from EDAM ontology [11]), example files (essential for testing purposes), default values and dependencies. This ensures human-readable documentation, FAIR metadata and programmatic accessibility can be generated consistently and comparably.
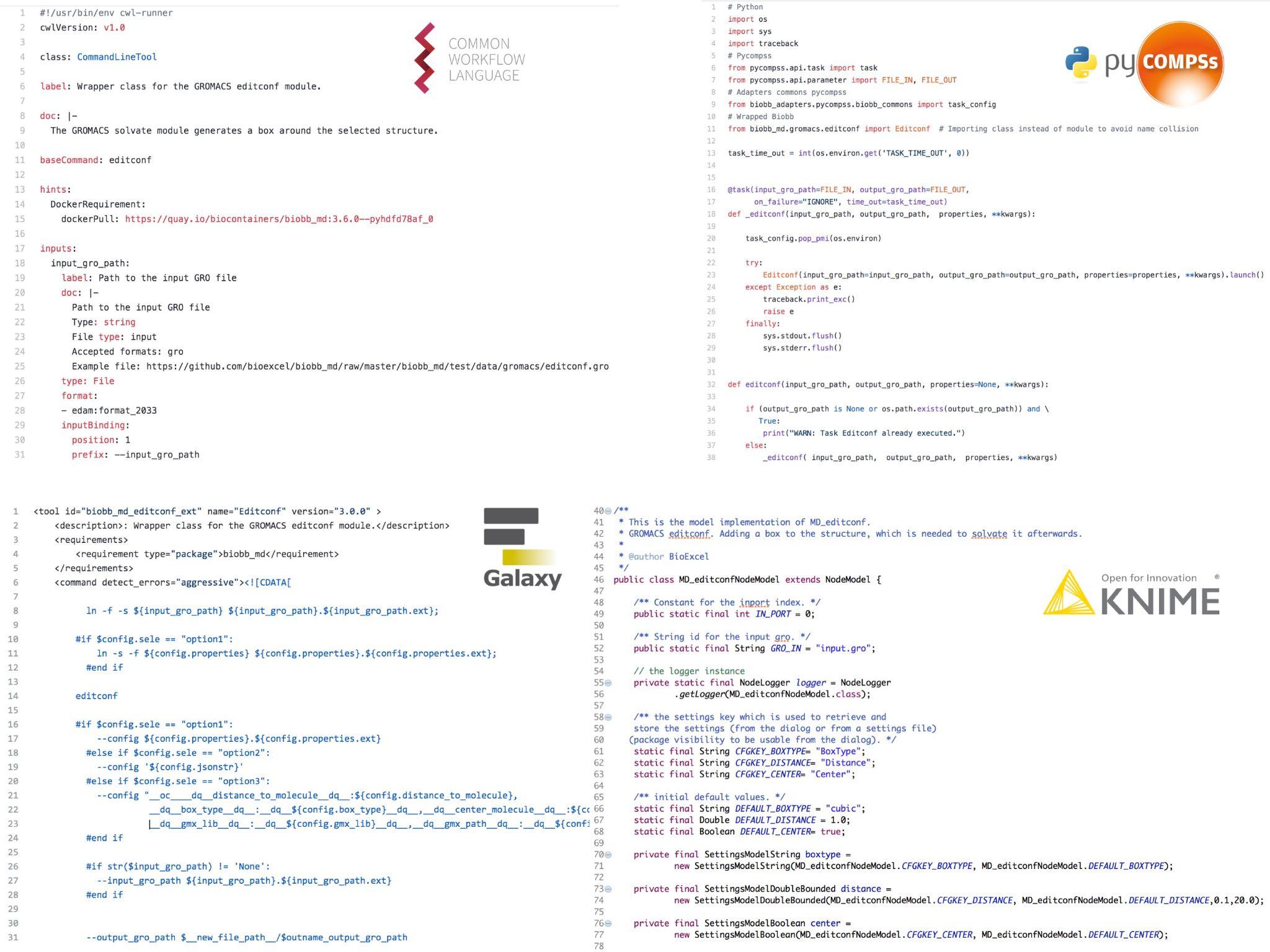
Code snippets for the BioBB WfMS bindings
CWL, PyCOMPSs, Galaxy and KNIME
The library is showcased through a collection of demonstration workflows [12]. Here, each workflow introduces individual building blocks as needed to explain a particular scientific computational method. We primarily expose the workflows as Jupyter Notebooks [13], which has been highlighted as a valuable tool for reproducible scientific workflows [14]. This offers a graphical interactive interface, including documentation (integrated markdown) related to the workflow and the building blocks used, but also to the biomolecular simulation methods used in the pipeline. Moreover, as we have demonstrated with our own Binder [15] hosting, these workflows are reproducible across platforms, assisted by BioConda [16] packaging of the building blocks and their software dependencies.
This assembly of available demonstration workflows have been successfully used in the BioExcel CoE for dissemination with a range of training events (e.g. BioExcel Summer & Winter School, webinars and virtual training). In training we particularly utilised the Binder infrastructure of the BioExcel Cloud portal [17] to give users a web-based first experience of the building blocks before they try them in other workflow systems.
We can observe that workflow building blocks such as BioBB are necessarily composed of a comprehensive list of digital objects, encompassing source code, packaging, containerization, documentation, attributions, citations, registry entries, WfMS integrations and REST APIs.
We propose to consider building blocks as composite digital objects in their own right: gathering the above software components along with their metadata, identifiers and operations then forms a Canonical Workflow Building Block (CWBB). We suggest this concept as a fundamental element of FAIR Digital Objects for Computational Workflows: researchers use the building blocks computationally as functional operations across WfMSs, while the FAIR aspect of CWBB propagates information and resources that are essential for reproducibility, reuse and understanding by anyone discovering the workflow.
2.1 Interoperability across different workflow languages
The concept of Canonical Workflow Building Blocks is here showcased with the BioBB library, by using a transversal workflow present in many different computational biomolecular projects: a Molecular Dynamics (MD) protein setup. This workflow prepares a protein structure to be used as input for an MD simulation, going through a series of steps where the protein is completed (adding hydrogen and missing atoms), optionally introducing a residue mutation, then submerging the protein in a virtual box of water molecules with a particular ionic concentration, and finally energetically equilibrating the system (so that solvent and ions are well accommodated around the protein at the desired temperature).
This simulation process involves a non-negligible number of steps, using a variety of biomolecular tools. The BioBB library was used to assemble this workflow, interconnecting building blocks using Python functions (Jupyter Notebook, Command Line Interface), auto-generated bindings (Galaxy [18], CWL [19], PyCOMPSs [20]) or manually generated bindings (KNIME [21]). Corresponding workflows for the different WfMS can be found in WorkflowHub [22,23,24,25,26] and graphical extracts can be seen in Figure 2.
This example demonstrates how the same canonical building blocks can be used in different WfMS. Wrappers and tools executed behind the workflows are exactly the same, but the workflows are built using different WfMS, some of them in a graphical way (drag & drop, Galaxy, KNIME), some in a command line way (Jupyter Notebook, PyCOMPSs, CWL); workflows can be focused on short/interactive executions (Jupyter Notebook), or on High Throughput/High Performance Computing (HT-HPC) executions (PyCOMPSs); some of them prepared for a particular WfMS installation (Galaxy), others completely system-agnostic (CWL).
The current number of available WfMS bindings include Jupyter Notebook, PyCOMPSs, CWL, Galaxy and KNIME WfMS, in addition to a command line mechanism. Thanks to the extensive documentation added in the source code as Python docstrings, new bindings for available WfMS can be generated. We are also experimenting with generating a REST API exposing the building services as Web services. However, it should be noted that such automatic generation of bindings is not always practically feasible. As an example, KNIME nodes require a complete Java skeleton code, as well as a definition of new data types for all inputs/outputs required, which makes their automatic generation a heavy and potentially error-prone task. Bindings for workflow languages with a domain-specific language (DSL) for tool definitions (e.g. Galaxy, CWL) can on the other hand be generated in a more straightforward fashion.
![Assembled in with 5 different workflow managers using BioBB canonical building blocks. From top-left: Galaxy [[22]], KNIME [[23]], CWL [[24]], Jupyter Notebook [[25]], PyCOMPSs [[26]].](figure2.png)
Protein MD Setup transversal workflow
Assembled in with 5 different workflow managers using BioBB canonical building blocks. From top-left: Galaxy [[22]], KNIME [[23]], CWL [[24]], Jupyter Notebook [[25]], PyCOMPSs [[26]].
The transversal protein MD setup workflow was chosen as a real example that is readily understandable by domain experts. More complex pipelines involving a broader set of wrapped biomolecular tools have been developed using the BioBB library, primarily as Jupyter Notebooks. A selection of these have similarly been assembled for different WfMS using the auto-generated bindings and uploaded to the WorkflowHub repository.
3. Discussion
Early work on libraries of workflows fragments include Web Service-based approaches where tools are wrapped and exposed using common, interoperable data types in BioMoby [27] for bioinformatics and similarly caBIG [28] for cancer genomics. While these efforts were interoperable across WfMSs they required a large up-front investment in agreeing to and adapting native data to common RDF or XML representations.
The notion of abstract workflows [29], structural workflow descriptions separated from their concrete execution realisations and augmented with Linked Data annotations, have been emphasised as essential for reuse and consistency across workflow systems. Identifying common motifs for workflow operations [30] (e.g. Data preparation, Format transformation, Filter, Combine) are important to simplify and understand otherwise fine-grained workflow provenance traces.
Most other efforts to standardise a set of disparate analytical tools have been done within the scope of a single WfMS, allowing customised user interaction, data visualisation, configuration and findability, for instance Taverna components had prototypical building blocks [31] which were instantiated at runtime by reference from a registry. KNIME components and metanodes, shared on the KNIME Hub are frequently designed to be interoperable, but with a perhaps weaker notion of component families. The Galaxy toolshed [32] is likewise populated with different sets of tool wrappers that are largely made to be interoperable within a category.
The Common Workflow Language (CWL) [19] has a strong emphasis on interoperable command line tool descriptions, with support for containers and Conda packaging, as well as support for FAIR metadata like contributors, license and EDAM ontology type annotations. With multiple leading workflow engines now supporting CWL, and experimental Galaxy support, this seems perhaps the most promising candidate for both making and describing canonical workflow building blocks, however we’ve identified a few stumbling blocks.
One obvious challenge is that the implementing WfMS needs to have CWL support, along with support for either containers or Conda packaging to find the described executables. While it is possible to run a CWL tool directly using a #!/usr/bin/env cwl-runner shebang on POSIX systems, this still requires pre-installation and possibly configuration of a CWL engine like cwltool or Toil [33]. However workflow engines have multiple dependencies and often cannot easily be run from a container themselves1.
Within the CWL community it was originally envisioned that a wider set of workflow systems would adopt CWL for tool description/execution, with a subset implementing full CWL workflow support. This would allow shared community effort for describing tools, say in the Common Workflow Library, rather than each WfMS needing to duplicate this tool wrapping in separate repositories and languages. However, with the exception of experimental tool support in Galaxy, in practice all CWL implementers have gone for full workflow support.
Another challenge is that making a set of building blocks frequently requires the use of shims, for instance file conversion, small search/replace operations or file renames. In a CWL approach these can either be performed with an Expression using JavaScript snippets which only has limited access to file content, or as an additional workflow step added before or after the main tool step. This combination could then be nested as a subworkflow, similar to KNIME’s metanodes, and would also be flexible by allowing different containers or packages for any pre- or post-steps. Such a CWL building block however becomes harder to access from a non-CWL WfMS, because of lack of control over configuration/execution options for the now nested CWL tools. In practice2, executing a nested CWL workflow from a native WfMS language would require the engine to implement full CWL Workflow support (or delegate to a CWL engine).
For the main BioBB building blocks we implemented demonstrator workflows that highlight how the tools should be used in different workflow management systems; each having a primary exemplar using Jupyter Notebook, which can be explored interactively using the BioExcel Binder. If we consider the abstract demonstrator workflows as canonical workflows they are therefore very much active objects, but can also be seen as workflow templates, as any real use case will need to specialise the workflow to tweak parameters, data selection etc.
We therefore also provide such workflow templates for multiple WfMS, including CWL, PyCOMPSs and Galaxy. These are fairly disparate workflow languages, yet by the use of the same canonical workflow building blocks (which again invoke the same software binaries), such WfMS-specific workflows effectively are instantiations of the same canonical workflow.
One challenge found is how to publish such canonical workflows in registries like the WorkflowHub. The hub supports the registration of Digital Objects in the form of RO-Crate [34], with the option of abstract CWL for describing the canonical workflow template, along with direct references to the workflow’s GitHub repository.
For instance in the RO-Crate for https://doi.org/10.48546/workflowhub.workflow.200.1 [26], which can also be rendered from GitHub, we have an entry for the main workflow according to the Workflow RO-Crate profile, detailing each canonical workflow building block used (e.g. biobb-md metadata). Here the FAIR aspect of the building blocks to help software citation is exercised, as the building block wrapper has one set of authors, documentation and licence (Apache-2.0), while the wrapped software (e.g. GROMACS metadata) has different authors, licence (GPL-2.1+) and documentation.
However, the deposit of such RO-Crates in WorkflowHub results in one registration entry per workflow language, which are not otherwise related and may not even share the same source code repository. Thus, we’ve identified the need for adding an overall canonical workflow entry, which can bring in workflow documentation and references shared across WfMS implementations, including a set of links to the more granular canonical workflow building blocks used by the workflow, but also to the individual WfMS implementations as separate digital objects.
A similar question of granularity applies at the workflow tool level [4], particularly for Findability and Accessibility, as we can consider at lowest granularity the scientific method in general (e.g. any algorithm for sequence alignment), followed by an application suite (bio.tools entry [35], homepage, documentation), instantiated as a particular software installation (Debian package, Docker container) with its dependencies at same level. The installation includes one or more software executables (a particular binary, a running service service), providing at the highest detailed granularity level the specific types of software functionality (a particular mode of operation, choice of analysis), for instance using certain command line flags.
For canonical workflow building blocks, with a focus on pluggable composability, this is mainly defined at this high granularity level of specific software functionality: explicit operations from an installed tool, which are then combined in a workflow. This is indeed the level WfMS tool definitions are typically done, e.g. a CWL Command Line Tool specifies a particular way to run a particular software binary. However, to be an actionable CWBB, the building block needs to additionally convey the lower granularity levels; particularly to support multiple options for interoperable installation and execution, as well as metadata at the most general level, such as documentation and scholarly citations.
While workflow management systems typically only operate at the highest granularity levels for execution details, and are frequently unaware of (or not exposing metadata at) the more general levels, we argue that in order for a Canonical Workflow [36] to follow and support FAIR principles for itself and its data, the workflow management system need to propagate structured metadata about the tools used by the workflow. We propose that in order to support the workflow’s applicability to multiple WfMS, the tools themselves must also have a consistent packaging and formal description that enables consistent computational invocation.
At the most general level, a canonical workflow built using such CWBBs is even conceptually reproducible because the FAIR documentation of the workflow, through its canonical workflow building blocks, identifies how individual tools and software applications are composed, which in worst case can be rebuilt using different installation methods in a different WfMS, or in best case inspected to detect and cross-link the same canonical workflow appearing in different WfMS instantiations. This view of software as composition of other software typically also applies at individual tool level, which themselves depend on programming language runtimes, libraries, services and reference data.
4. Requirements for Canonical Workflow Building Blocks
Building on the experiences with BioBB, we here propose requirements and recommendations for establishing Canonical Workflow Building Blocks (CWBB) as implementations of canonical steps introduced for Canonical Workflow Frameworks for Research [36].
The core purpose of a CWBB is to wrap a command line tool or other software that can perform an operation as part of a computational workflow. As such, the general advice for making software workflow-ready applies [37] (e.g. easy to install, documented, parallelizable, reproducible output), however a CWBB is also permitted to make use of additional scripts or shims to further adapt a third-party tool for workflow use and for data interoperability across blocks.
The way tools are installed or invoked varies slightly across WfMS and operating systems, therefore a CWBB should provide multiple methods for distributing software; currently containers (Docker, Singularity) and distribution-independent packaging (e.g. Conda, Homebrew) are promising by having reproducible install recipes and a wide range of open source dependencies (e.g. Java, Python). Additionally building blocks should allow overriding execution paths, e.g. for use with HPC module system and hardware-optimised binaries.
The CWBBs should have sufficient annotations to be able to generate bindings for different WfMSs and REST APIs, e.g. parameter names and descriptions, types and default values; enumerators for options, file formats for inputs/outputs.
Building blocks should be grouped into families that are interoperable through common data structures and file formats, as well as having joint naming conventions for configuration options. A CWBB family should be released as a single version following semantic versioning rules, which should have a corresponding persistent identifier (PID) [38].
Metadata for CWBBs should be captured following FAIR guidelines, and distributed as part of the block family and resolvable from the PID as a FAIR Digital Object. Metadata should include references to the CWBB software distributions (e.g. quay.io container URL) as well as attributions, citations and documentation for the wrapped tool.
Example workflows showing CWBB usage should be included in a WfMS-neutral language such as Jupyter Notebooks, which may have equivalent variants for each workflow binding. These workflows should be registered in a workflow registry like WorkflowHub or Dockstore, and assigned their own PIDs.
5. Conclusions
The proposed concept of Canonical Workflow Building Blocks can bridge the gap between FAIR Computational Workflows, interoperable reproducibility and for building canonical workflow descriptions to be used and described FAIRly across WfMSs.
The realisation of CWBBs can be achieved in many ways, not necessarily using the Python programming language together with RO-Crate as explored here. In particular if the envisioned Canonical Workflow Frameworks for Research become established in multiple WfMSs with the use of FAIR Digital Objects, the different implementations will need to agree on object types, software packaging and metadata formats in order to reuse tools and provide interoperable reproducibility for canonical workflows.
Likewise, to build a meaningful collection of building blocks for a given research domain, a directed collaborative effort is needed to consistently wrap tools for a related set of WfMSs, chosen to target particular use cases (a family of canonical workflows).
For individual users, a library of Canonical Workflow Building Blocks simplifies many aspects of building pipelines, beyond the FAIR aspects and data compatibility across blocks. For instance, they can benefit from training of a CWBB family using Jupyter Notebooks, and then use this knowledge to utilise the same building blocks in a scalable HPC workflow with a CWL engine like Toil, knowing they will perform consistently thanks to the use of containers.
While we have demonstrated CWBB in the biomedical domain, this approach is generally applicable to a wide range of sciences that execute pipelines of multiple file-based command line tools, however it may be harder to achieve with more algebraic “in memory” types of computational workflows, where steps could be challenging to containerize and distinguish as separate block.
We admit that biomolecular research is quite a homogenous field with respect to computational analyses and now becoming relatively mature in terms of tool composability in workflows, building on the experiences of the “FAIR pioneers” in the field of bioinformatics. Other fields, such as social sciences or ecology, can have a wider variety of methods and computational tools, often with human interactions, and may have to adapt the software to be workflow-ready [37] before using them as Canonical Workflow Building Blocks. Domains adapting CWBB approach (or workflow systems in general) should take note of the great benefits of hosting collaborative events where developers meet each other and their potential users, demonstrated in our field with events such WorkflowsRI [39] and Biohackathons [40].
The Common Workflow Language shows promise as a general canonical workflow building blocks mechanism: gathering execution details of tools along with their metadata and references, augmented with abstract workflows to represent canonical workflows. However, this would need further work to implement our CWBB recommendations in full. Future work for the Canonical Workflow Building Blocks concept includes formalising and automating publication practises, to make individual blocks available as FAIR Digital Objects on their own or as part of an aggregate collection like RO-Crate.
Acknowledgements
This work has been done as part of the BioExcel CoE (https://www.bioexcel.eu/), a project funded by the European Union contracts H2020-INFRAEDI-02-2018 823830, H2020-EINFRA-2015-1 675728. Additional work is funded through EOSC-Life (https://www.eosc-life.eu/) contract H2020-INFRAEOSC-2018-2 824087, and ELIXIR-CONVERGE (https://elixir-europe.org/) contract H2020-INFRADEV-2019-2 871075.
Author contributions
Author contributions according to the CASRAI CRediT taxonomy:
- Stian Soiland-Reyes
- Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Writing – original draft, Writing – review & editing
- Genís Bayarri
- Software, Software Documentation
- Pau Andrio
- Methodology, Software, Validation, Software Documentation
- Robin Long
- Software, Software Documentation
- Douglas Lowe
- Software, Software Documentation
- Ania Niewielska
- Methodology, Resources, Software
- Adam Hospital
- Methodology, Project administration, Resuorces, Software, Validation, Visualization, Writing – original draft, Writing – review & editing
- Paul Groth
- Methodology, Supervision, Writing – review & editing
The authors would also like to acknowledge contributions from: Felix Amaladoss, Cibin Sadasivan Baby, Finn Bacall, Rosa M. Badia, Sarah Butcher, Gerard Capes, Michael R. Crusoe, Alberto Eusebi, Carole Goble, Josep Lluís Gelpí, Modesto Orozco, Geoff Williams, Felix Amaladoss
References
[1] Victoria Stodden, Marcia McNutt, David H. Bailey, Ewa Deelman, Yolanda Gil, Brooks Hanson, Michael A. Heroux, John P.A. Ioannidis, Michela Taufer (2016):
Enhancing reproducibility for computational methods.
Science 354(6317) pp 1240–1241
https://doi.org/10.1126/science.aah6168
[2] Jeremy Leipzig, Daniel Nüst, Charles Tapley Hoyt, Karthik Ram, Jane Greenberg (2021):
The role of metadata in reproducible computational research. Patterns 2(9):100322.
https://doi.org/10.1016/j.patter.2021.100322
[3] Daniel S. Katz, Morane Gruenpeter, Tom Honeyman, Lorraine Hwang, Mark D. Wilkinson, Vanessa Sochat, Hartwig Anzt, Carole Goble, FAIR4RS Subgroup 1 (2021):
A Fresh Look at FAIR for Research Software.
arXiv:2101.10883
[4] Steffen Möller, Stuart W. Prescott, Lars Wirzenius, Petter Reinholdtsen, Brad Chapman, Pjotr Prins, Stian Soiland-Reyes, Fabian Klötzl, Andrea Bagnacani, Matúš Kalaš, Andreas Tille, Michael R. Crusoe (2017):
Robust cross-platform workflows: How technical and scientific communities collaborate to develop, test and share best practices for data analysis. Data Science and Engineering 2 pp 232–244.
https://doi.org/10.1007/s41019-017-0050-4
[5] Sarah Cohen-Boulakia, Khalid Belhajjame, Olivier Collin, Jérôme Chopard, Christine Froidevaux, Alban Gaignard, Konrad Hinsen, Pierre Larmande, Yvan Le Bras, Frédéric Lemoine, Fabien Mareuil, Hervé Ménager, Christophe Pradal, Christophe Blanchet (2017):
Scientific Workflows for Computational Reproducibility in the Life Sciences: Status, Challenges and Opportunities. Future Generation Computer Systems 75 pp 284–298.
https://doi.org/10.1016/j.future.2017.01.012
[6] Björn Grüning, John Chilton, Johannes Köster, Ryan Dale, Nicola Soranzo, Marius van den Beek, Jeremy Goecks, Rolf Backofen, Anton Nekrutenko, James Taylor (2018):
Practical Computational Reproducibility in the Life Sciences. Cell Systems 6(6) pp 631–635.
https://doi.org/10.1016/j.cels.2018.03.014
[7] Anna-Lena Lamprecht, Leyla Garcia, Mateusz Kuzak, Carlos Martinez, Ricardo Arcila, Eva Martin Del Pico, Victoria Dominguez Del Angel, et al. (2020):
Towards FAIR Principles for Research Software. Data Science 3(1) pp 37–59.
https://doi.org/10.3233/ds-190026
[8] Koenraad De Smedt, Dimitris Koureas, Peter Wittenburg (2020):
FAIR Digital Objects for Science: From Data Pieces to Actionable Knowledge Units.
Publications 8(2):21
https://doi.org/10.3390/publications8020021
[9] Carole Goble, Sarah Cohen-Boulakia, Stian Soiland-Reyes, Daniel Garijo, Yolanda Gil, Michael R. Crusoe, Kristian Peters, Daniel Schober (2020):
FAIR Computational Workflows. Data Intelligence 2(1) pp 108–121.
https://doi.org/10.1162/dint_a_00033
[10] Pau Andrio, Adam Hospital, Javier Conejero, Luis Jordá, Marc Del Pino, Laia Codo, Stian Soiland-Reyes, Carole Goble, Daniele Lezzi, Rosa M. Badia, Modesto Orozco, Josep Ll. Gelpi (2019):
BioExcel Building Blocks, a software library for interoperable biomolecular simulation workflows. Scientific Data 6:169
https://doi.org/10.1038/s41597-019-0177-4
[11] Jon Ison, Matúš Kalaš, Inge Jonassen, Dan Bolser, Mahmut Uludag, Hamish McWilliam, James Malone, Rodrigo Lopez, Steve Pettifer, Peter Rice (2013):
EDAM: an ontology of bioinformatics operations, types of data and identifiers, topics and formats. Bioinformatics 29(10) pp 1325–1332.
https://doi.org/10.1093/bioinformatics/btt113
[12] Adam Hospital, Genís Bayarri, Stian Soiland-Reyes, Jose Lluis Gelpi, Pau Andrio, Daniele Lezzi, Sarah Butcher, Ania Niewielska, Yvonne Westermaier, Rosa Maria Badia, Rodrigo Vargas, Alexandre Bonvin (2020):
BioExcel-2 Deliverable 2.3 – First release of demonstration workflows. Project deliverable, Zenodo.
https://doi.org/10.5281/zenodo.4540432
[13] Thomas Kluyver, Benjamin Ragan-Kelley, Fernando Pérez, Brian Granger, Matthias Bussonnier, Jonathan Frederic, Kyle Kelley, Jessica Hamrick, Jason Grout, Sylvain Corlay, Paul Ivanov, Damián Avila, Safia Abdalla, Carol Willing, Jupyter Development Team (2016):
Jupyter Notebooks – a publishing format for reproducible computational workflows. Positioning and Power in Academic Publishing: Players, Agents and Agendas pp 87 - 90. Fernando Loizides, Birgit Schmidt (eds), Proceedings of the 20th International Conference on Electronic Publishing.
https://doi.org/10.3233/978-1-61499-649-1-87
[14] Marijan Beg, Juliette Taka, Thomas Kluyver, Alexander Konovalov, Min Ragan-Kelley, Nicolas M. Thiery, Hans Fangohr (2021):
Using Jupyter for Reproducible Scientific Workflows. Computing in Science & Engineering 23(2) pp 36–46.
https://doi.org/10.1109/mcse.2021.3052101
[15] Jupyter Project, Matthias Bussonnier, Jessica Forde, Jeremy Freeman, Brian Granger, Tim Head, Chris Holdgraf, et al. (2018):
Binder 2.0 - Reproducible, Interactive, Sharable Environments for Science at Scale. Proceedings of the 17th Python in Science Conference. SciPy, 2018.
https://doi.org/10.25080/majora-4af1f417-011
[16] Björn Grüning, Ryan Dale, Andreas Sjödin, Brad A. Chapman, Jillian Rowe, Christopher H. Tomkins-Tinch, Renan Valieris, the Bioconda Team, Johannes Köster (2018):
Bioconda: Sustainable and Comprehensive Software Distribution for the Life Sciences. Nature Methods 15 pp 475–476.
https://doi.org/10.1038/s41592-018-0046-7.
[17] Ania Niewielska, Sarah Butcher, Yvonne Westermaier (2020):
BioExcel-2 Deliverable 2.5 - Provision of a Workflow Environment at BioExcel portal. Zenodo
https://doi.org/10.5281/zenodo.4916060
[18] Enis Afgan, Dannon Baker, Bérénice Batut, Marius van den Beek, Dave Bouvier, Martin Čech, John Chilton, Dave Clements, Nate Coraor, Björn Grüning, Aysam Guerler, Jennifer Hillman-Jackson, Vahid Jalili, Helena Rasche, Nicola Soranzo, Jeremy Goecks, James Taylor, Anton Nekrutenko, and Daniel Blankenberg (2018):
The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Research 46*(W1)* pp W537–W544.
https://doi.org/10.1093/nar/gky379
[19] Michael R. Crusoe, Sanne Abeln, Alexandru Iosup, Peter Amstutz, John Chilton, Nebojša Tijanić, Hervé Ménager, Stian Soiland-Reyes, Carole Goble, The CWL Community (2022):
Methods Included: Standardizing Computational Reuse and Portability with the Common Workflow Language. Communication of the ACM (in press). arXiv:2105.07028
https://doi.org/10.1145/3486897
[20] Enric Tejedor, Yolanda Becerra, Guillem Alomar, Anna Queralt, Rosa M. Badia, Jordi Torres, Toni Cortes, Jesús Labarta (2017):
PyCOMPSs: Parallel computational workflows in Python. The International Journal of High Performance Computing Applications 31(1) pp 66–82.
https://doi.org/10.1177/1094342015594678
[21] Alexander Fillbrunn, Christian Dietz, Julianus Pfeuffer, René Rahn, Gregory A. Landrum, Michael R. Berthold (2017):
KNIME for reproducible cross-domain analysis of life science data. Journal of Biotechnology 261 pp 149–156.
https://doi.org/10.1016/j.jbiotec.2017.07.028
[22] Douglas Lowe (2021):
Protein MD Setup tutorial using BioExcel Building Blocks (biobb) in Galaxy. WorkflowHub. Workflow (Galaxy).
https://doi.org/10.48546/workflowhub.workflow.194.1
[23] Adam Hospital (2021):
Protein MD Setup tutorial using BioExcel Building Blocks (biobb) in KNIME. WorkflowHub. Workflow (KNIME).
https://doi.org/10.48546/workflowhub.workflow.201.1
[24] Genís Bayarri, Robin Long (2021):
Protein MD Setup tutorial using BioExcel Building Blocks (biobb) in CWL. WorkflowHub. Workflow (CWL).
https://doi.org/10.48546/workflowhub.workflow.29.3
[25] Genís Bayarri, Adam Hospital, Douglas Lowe (2021):
Protein MD Setup tutorial using BioExcel Building Blocks (biobb) in Jupyter Notebook. WorkflowHub. Workflow (Jupyter Notebook).
https://doi.org/10.48546/workflowhub.workflow.120.2
[26] Adam Hospital, Pau Andrio (2021):
Protein MD Setup HPC tutorial using BioExcel Building Blocks (biobb) in PyCOMPSs. WorkflowHub. Workflow (PyCOMPSs).
https://doi.org/10.48546/workflowhub.workflow.200.1
[27] The BioMoby Consortium (2008):
Interoperability with Moby 1.0—It’s better than sharing your toothbrush! Briefings in Bioinformatics 9(3) pp 220–231.
https://doi.org/10.1093/bib/bbn003
[28] Joel Saltz, Scott Oster, Shannon Hastings, Stephen Langella, Tahsin Kurc, William Sanchez, Manav Kher, Arumani Manisundaram, Krishnakant Shanbhag, Peter Covitz (2006):
caGrid: design and implementation of the core architecture of the cancer biomedical informatics grid. Bioinformatics 22(15) pp 1910–1916.
https://doi.org/10.1093/bioinformatics/btl272
[29] Daniel Garijo, Yolanda Gil (2011):
A New Approach for Publishing Workflows. Proceedings of the 6th Workshop on Workflows in Support of Large-Scale Science - WORKS ‘11.
https://doi.org/10.1145/2110497.2110504
[30] Daniel Garijo, Pinar Alper, Khalid Belhajjame, Oscar Corcho, Yolanda Gil, Carole Goble (2014):
Common Motifs in Scientific Workflows: An Empirical Analysis. Future Generation Computer Systems 36 pp 338–351.
https://doi.org/10.1016/j.future.2013.09.018
[31] Renato De Giovanni, Alan R. Williams, Vera Hernández Ernst, Robert Kulawik, Francisco Quevedo Fernandez, Alex R. Hardisty (2016):
ENM Components: a new set of web service‐based workflow components for ecological niche modelling. Ecography 39(4) pp 376–383.
https://doi.org/10.1111/ecog.01552
[32] Daniel Blankenberg, Gregory Von Kuster, Emil Bouvier, Dannon Baker, Enis Afgan, Nicholas Stoler, James Taylor, Anton Nekrutenko, the Galaxy Team (2014):
Dissemination of scientific software with Galaxy ToolShed. Genome Biology 15:403
https://doi.org/10.1186/gb4161
[33] John Vivian, Arjun Arkal Rao, Frank Austin Nothaft, Christopher Ketchum, Joel Armstrong, Adam Novak, Jacob Pfeil, Jake Narkizian, Alden D Deran, Audrey Musselman-Brown, Hannes Schmidt, Peter Amstutz, Brian Craft, Mary Goldman, Kate Rosenbloom, Melissa Cline, Brian O’Connor, Megan Hanna, Chet Birger, W James Kent, David A Patterson, Anthony D Joseph, Jingchun Zhu, Sasha Zaranek, Gad Getz, David Haussler & Benedict Paten (2017):
Toil enables reproducible, open source, big biomedical data analyses. Nature Biotechnology 35 pp 314–316.
https://doi.org/10.1038/nbt.3772
[34] Stian Soiland-Reyes, Peter Sefton, Mercè Crosas, Leyla Jael Castro, Frederik Coppens, José M. Fernández, Daniel Garijo, Björn Grüning, Marco La Rosa, Simone Leo, Eoghan Ó Carragáin, Marc Portier, Ana Trisovic, RO-Crate Community, Paul Groth, Carole Goble (2022):
Packaging research artefacts with RO-Crate. Data Science (pre-press).
https://doi.org/10.3233/DS-210053
[35] Jon Ison, Hans Ienasescu, Emil Rydza, Piotr Chmura, Kristoffer Rapacki, Alban Gaignard, Veit Schwämmle, Jacques van Helden, Matúš Kalaš, Hervé Ménager (2021):
biotoolsSchema: a formalized schema for bioinformatics software description. GigaScience, 10(1):giaa157
https://doi.org/10.1093/gigascience/giaa157
[36] The CWFR Group, Alex Hardisty (ed.), Peter Wittenburg (ed.) (2021):
CWFR Position Paper. OSF, January 6, 2021.
https://osf.io/3rekv/
[37] Paul Brack, Peter Crowther, Stian Soiland-Reyes, Stuart Owen, Douglas Lowe, Alan R Williams, Quentin Groom, Mathias Dillen, Frederik Coppens, Björn Grüning, Ignacio Eguinoa, Phil Ewels, Carole Goble (2022):
10 Simple Rules for making a software tool workflow-ready. PLOS Computational Biology (accepted), preprint PCOMPBIOL-D-21-01704R1 at https://zenodo.org/record/5901220
[38] Julie A McMurry, Nick Juty, Niklas Blomberg, Tony Burdett, Tom Conlin, Nathalie Conte, Mélanie Courtot, John Deck, Michel Dumontier, Donal K Fellows, Alejandra Gonzalez-Beltran, Philipp Gormanns, Jeffrey Grethe, Janna Hastings, Jean-Karim Hériché, Henning Hermjakob, Jon C Ison, Rafael C Jimenez, Simon Jupp, John Kunze, Camille Laibe, Nicolas Le Novère, James Malone, Maria Jesus Martin, Johanna R McEntyre, Chris Morris, Juha Muilu, Wolfgang Müller, Philippe Rocca-Serra, Susanna-Assunta Sansone, Murat Sariyar, Jacky L Snoep, Stian Soiland-Reyes, Natalie J Stanford, Neil Swainston, Nicole Washington, Alan R Williams, Sarala M Wimalaratne, Lilly M Winfree, Katherine Wolstencroft, Carole Goble, Cristopher J Mungall, Melissa A Haendel, Helen Parkinson (2017):
Identifiers for the 21st century: How to design, provision, and reuse identifiers to maximize utility and impact of life science data. PLOS Biology 15(6):e2001414
https://doi.org/10.1371/journal.pbio.2001414
[39] Rafael Ferreira da Silva, Henri Casanova, Kyle Chard, Ilkay Altintas, Rosa M Badia, Bartosz Balis, Tainã Coleman, Frederik Coppens, Frank Di Natale, Bjoern Enders, Thomas Fahringer, Rosa Filgueira, Grigori Fursin, Daniel Garijo, Carole Goble, Dorran Howell, Shantenu Jha, Daniel S. Katz, Daniel Laney, Ulf Leser, Maciej Malawski, Kshitij Mehta, Loïc Pottier, Jonathan Ozik, J. Luc Peterson, Lavanya Ramakrishnan, Stian Soiland-Reyes, Douglas Thain, Matthew Wolf (2021):
A Community Roadmap for Scientific Workflows Research and Development.
arXiv:2110.02168 [cs.DC]
[40] Leyla Garcia, Erick Antezana, Alexander Garcia, Evan Bolton, Rafael Jimenez, Pjotr Prins, Juan M. Banda, Toshiaki Katayama (2020):
Ten simple rules to run a successful BioHackathon. PLOS Computational Biology 16(5):e1007808.
https://doi.org/10.1371/journal.pcbi.1007808
-
To execute the wrapped tool, a containerized workflow engine would need nested containers which are not generally recommended for security reasons. It is possible to work around this limitation using Singularity or Conda. ↩︎
-
It is worth mentioning that it would also be possible to generate WfMS-specific bindings from CWL descriptions (e.g. as demonstrated with cwl2script for Bash, gxargparse for Galaxy, cwl2wdl for WDL), although this necessitates constraining the tool and workflow definitions to a limited mappable subset of CWL. ↩︎